UCI-led study confirms linkage between altered DNA repair and DNA damage in neurodegenerative conditions causing debilitating movement disorders
Source: UCI School of Medicine
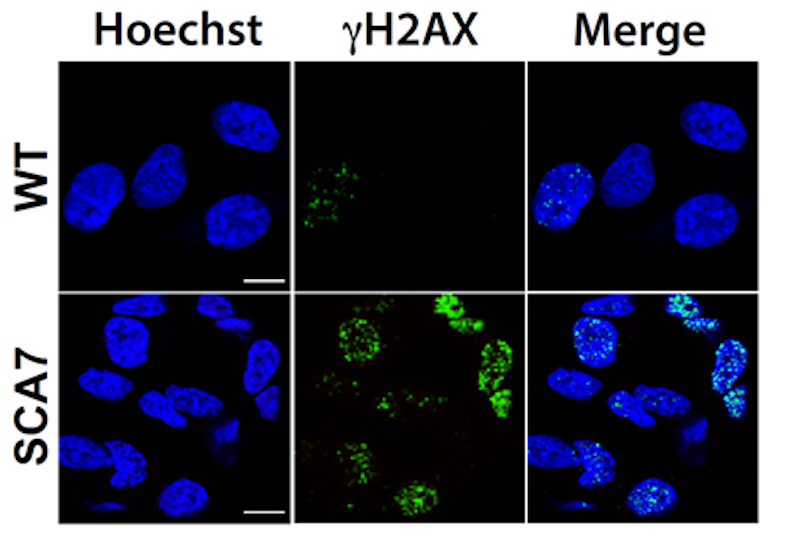
Increased DNA damage in neurons from SCA7 patients. Skin cells from an SCA7 patient and an unaffected sibling (WT), generated induced pluripotent stem cells. Those stem cells were used to make neurons which were grown in culture. Illustrated here are stained neurons with Hoechst to show the nucleus, and with ɣH2AX, which is a marker of DNA damage.
UCI School of Medicine
Irvine, Calif., Nov. 30, 2021 — A new study led by University of California, Irvine researchers has confirmed a link between altered DNA repair and increased DNA damage associated with spinocerebellar ataxia type 7 (SCA7), a debilitating, sometimes deadly neurodegenerative condition causing movement disorders. Their work also revealed a potential therapeutic target for the currently incurable and difficult to treat condition.
Titled, “Altered H3 histone acetylation impairs high-fidelity DNA repair to promote cerebellar degeneration in spinocerebellar ataxia type 7,” the study was published today in Cell Reports.
“Our research delves into the mechanistic basis of the cerebellar neuron degeneration and death in SCA7, a specific SCA that causes impaired coordination, like difficulty with walking, talking and eye movement,” said corresponding author Albert La Spada, Distinguished Professor of pathology, neurology and biological chemistry in the UCI School of Medicine. “Through our efforts, we confirmed the connection between altered DNA repair and DNA damage, and also the importance of the activation of PARP1 enzymes that results.”
Previous research has shown that increased DNA damage can lead to the activation of PARP1 enzymes. These enzymes serve to recruit the DNA repair machinery but can also promote cerebellar neuron dysfunction and death. Fortunately, PARP inhibitors already exist and could prove to be a promising potential new treatment.
“Perhaps more exciting is that the DNA damage and altered DNA repair found in SCA7, is also found in other SCAs. This could mean new therapeutics to treat the devastating effects of many of the forms of spinocerebellar ataxia might be possible,” said LaSpada. “Our next steps will be to test candidate PARP inhibitor drugs in mouse models of SCA7 as well as in neurons derived from pluripotent stem cells generated from human SCA7 human patients.”
SCA7, belongs to a disease category which includes spinobulbar muscular atrophy (SBMA), Huntington disease (HD), dentatorubral-pallidoluysian atrophy (DRPLA), and five other forms of spinocerebellar ataxia (SCA1, 2, 3, 6, and 17).
Each year, 15,000 - 20,000 Americans suffer from spinocerebellar ataxia (SCA), a genetic dominantly inherited neurodegenerative condition, that frequently results in atrophy of the cerebellum and the loss of fine coordination of muscle movements leading to unsteady and clumsy motion, and other symptoms. SCAs can affect anyone of any age. Currently, there are no known effective treatments or cures.
This work was supported by grants from the National Institutes of Health and the Polish Ministry of Science and Higher Education.
